Align reads with bowtie
Bowtie align reads on indexed genomes¶
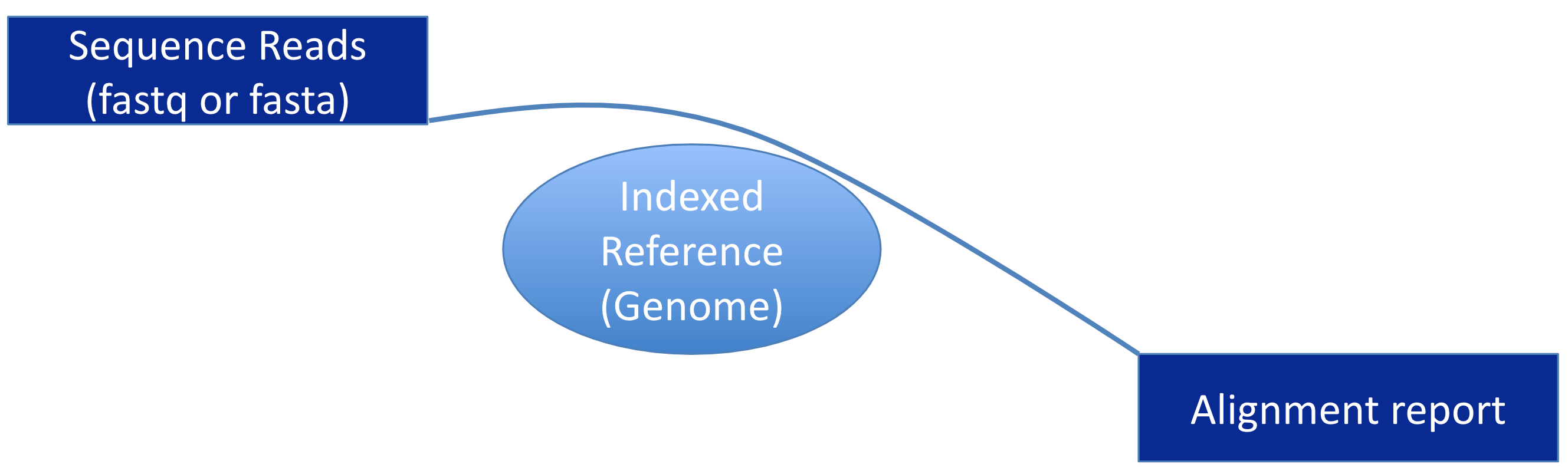
Preliminary Note  ¶
¶
For the following steps, you will need 2 programs which an admin (with admin rights) has already installed - system-wide - for you, using the following command:
Prepare dmel_r6.54 bowtie index (Drosophila genome)¶
 This step should take about 2-3 min
This step should take about 2-3 min
Align the clipped fasta reads to dmel.r6.54 using bowtie¶
bowtie dmel.r6.54 -f clipped_GRH-103_R1.fasta \
-v 0 \
-k 1 \
-p 3 \
--al dmel_matched_GRH-103.fa \
--un unmatched_GRH-103.fa \
-S > GRH-103.sam
The bowtie alignment command explained
bowtie dmel.r6.54 -f clipped_GRH-103_R1.fasta# tells bowtie where is the index and the input clipped_GRH-103_R1.fasta-v 0 -k 1 -p 3# These are bowtie options--al dmel_matched_GRH-103.fa# aligned reads will be in the dmel_matched_GRH-103.fa file--un unmatched_GRH-103.fa# Unaligned reads will be in the unmatched_GRH-103.fa file-S > GRH-103.sam# tells bowtie to return an alignement file in SAM format (-S) -S > GRH-103.sam