Compute the genome read coverage
To interpretate the likelihood of translocation calls, we need to know the sequencing depth at the breakpoints.
This can be done by computing the genome read coverage from the BAM alignments, using the tool:
bamCoverage generates a coverage bigWig file from a given BAM or CRAM file (Galaxy Version 3.1.2.0.0)
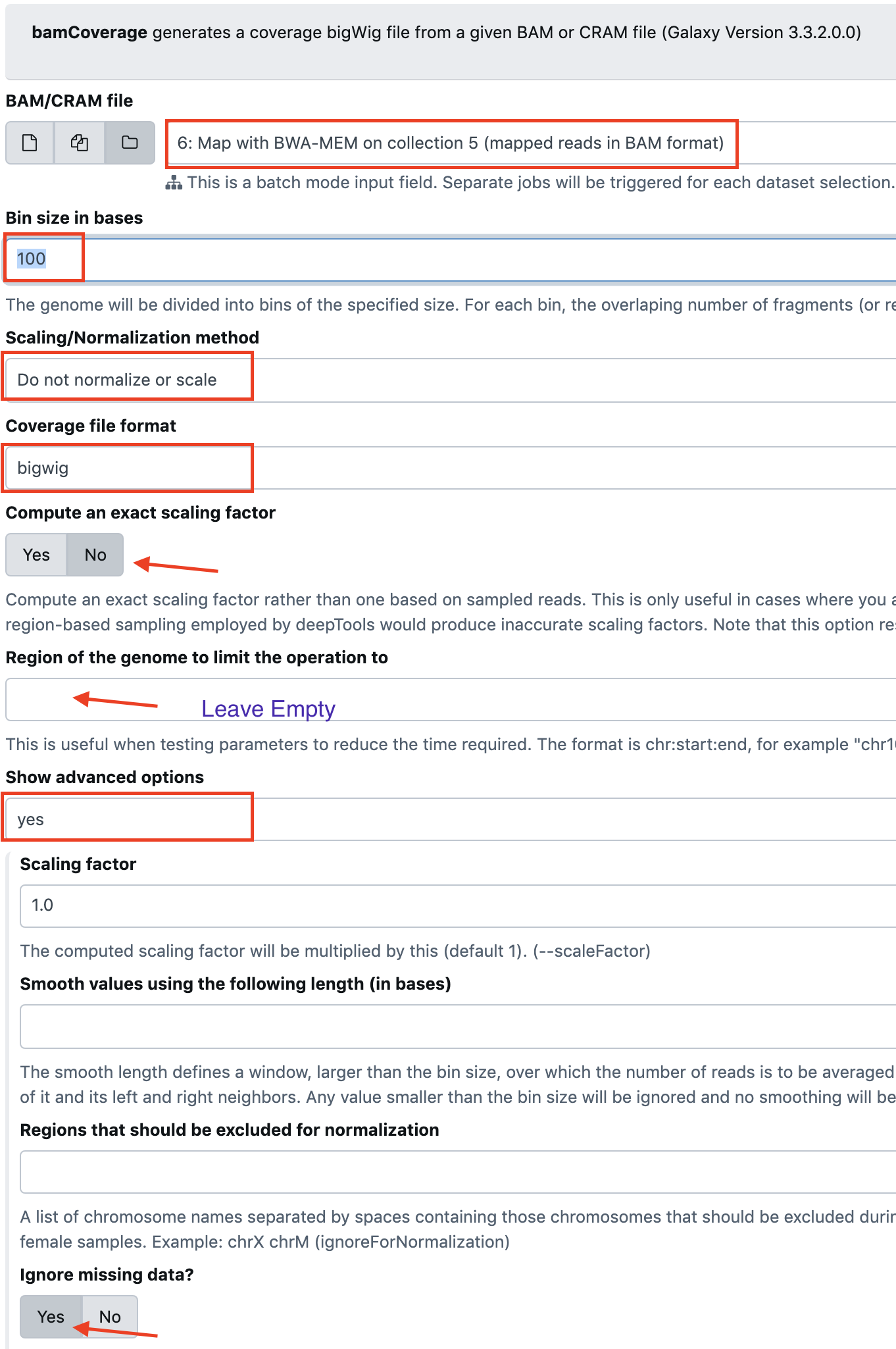
- BAM/CRAM file:
Dataset CollectionandMap with BWA-MEM on collection 3 (mapped reads in BAM format) - Bin size in bases:
100 - Scaling/Normalization method:
Do not normalize or scale - Coverage file format:
bigWig - Compute an exact scaling factor:
no - Region of the genome to limit the operation to: Leave empty
- Show advanced options:
yes - Ignore missing data?:
yes - Other options unchanged
- Run the tool