Counting strategy  ¶
¶
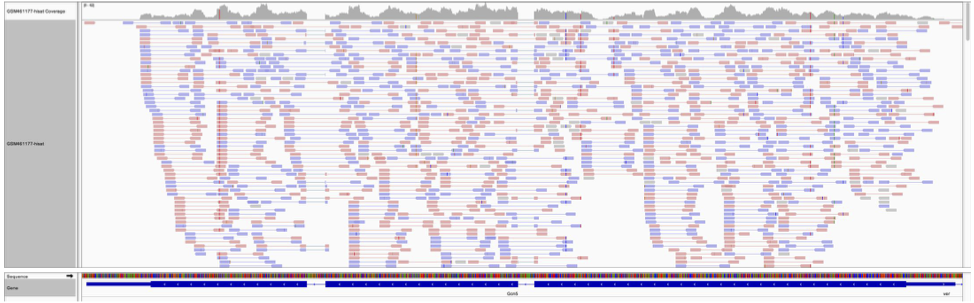
Count the number of reads per annotated gene¶
To compare the expression of single genes between different conditions the first step is to quantify the number of reads per gene.

From the image above, we can compute:
Number of reads per exons¶
| Gene | Exon | Number of reads |
|---|---|---|
| gene1 | exon1 | 3 |
| gene1 | exon2 | 2 |
| gene2 | exon1 | 3 |
| gene2 | exon2 | 4 |
| gene2 | exon3 | 3 |
- The gene1 has 4 reads, not 5 (gene1 - exon1 + gene1 - exon2) because of the splicing of the last read.
- The gene2 has 6 reads (3 spliced reads)
Counting tools¶
Two main tools could be used for that: HTSeq-count (Anders et al, Bioinformatics, 2015) or featureCounts (Liao et al, Bioinformatics, 2014).
FeatureCounts is considerably faster and requires far less computational resources.
HTSeq-counts was originally developed by Simon Anders (the developer of DESeq2 and DEXSeq). Thus, there is certainly a guaranty of quality. However, it is less easy to use and requires several additional steps, in particular to carefully control the GTF/GFF files taken as an input by HTSeq-counts.
Originally, FeatureCounts and and HTSEQq-counts returned similar but not identical genes counts. A few percents differences was noted in the original FeatureCount article (and see the figure below). This was likely due to the use a completely different algorithms to assign reads to the genomic features (either genes or exons).
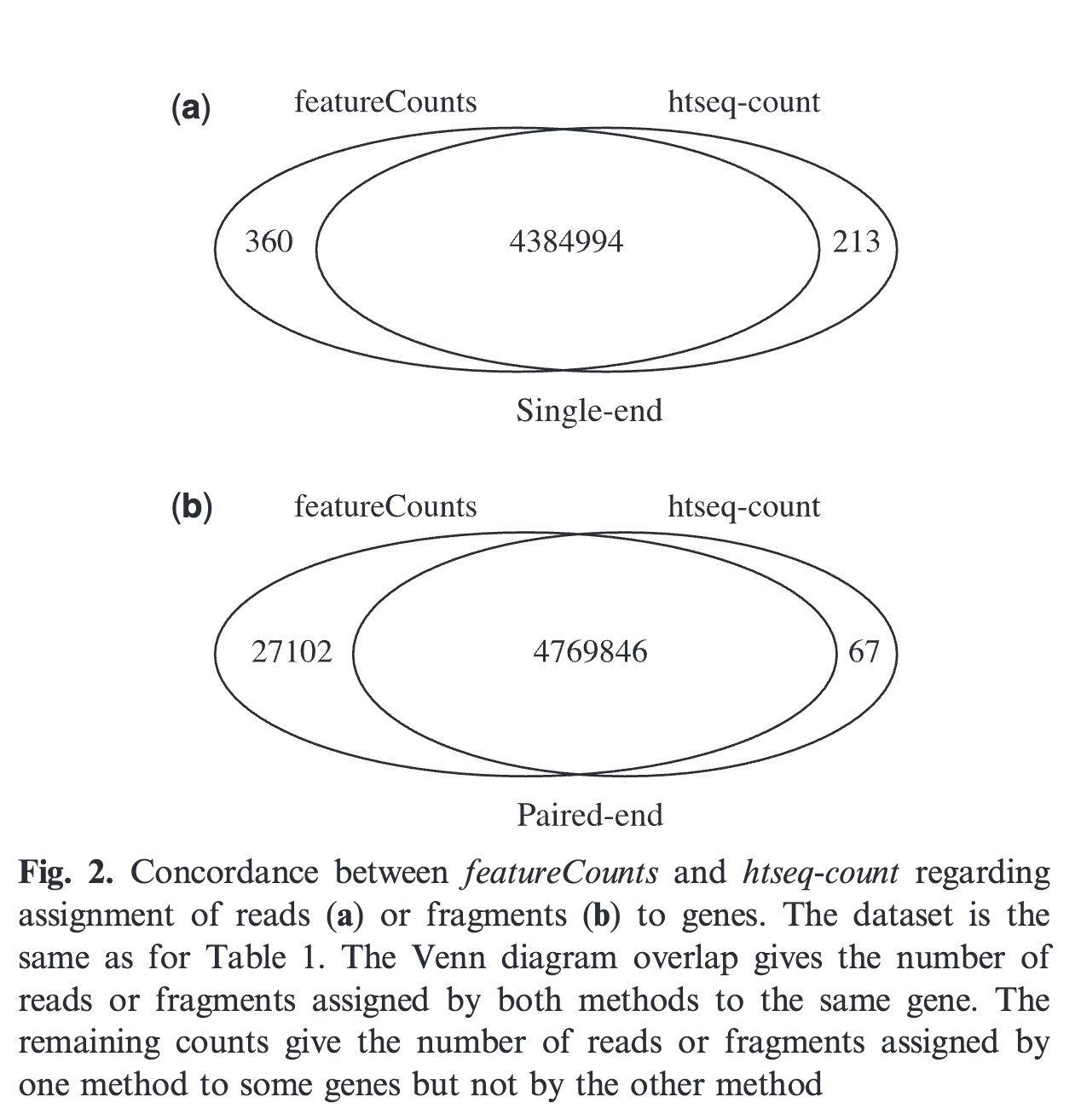
However, nowadays the 2 programs returns quasi-identical, if not identical, results.
In the next sections, you will have the occasion to give a shot to both them and see that counts returned are identical.
Three important points
- Here again (in the counting task), the genome annotation file, either a GTF or a GFF3, is central, regardless of the chosen counting software.
- The library strandness is absolutly required for an accurate gene read counting. In the case of library with reverse strandness, only sequences antisense to the gene transcripts will be counted. On the contrary, with forward strandness library, only sense reads will be counted. Finally, all reads will be counted in the case of unstranded libraries.
Importantly, note that in the case of genes overlapping on + and -
genomes strands, only stranded libraries (regardless of their strandness) will allow
to assign specifically reads to each of the genes. With unstranded libraries, these
overlapping genes are not counted, unless you implement a statistical model for inferring
the gene origin of the reads.
- If you are working with paired-end sequencing datasets, you will not count reads. Instead, you will count fragments. If you count reads in this situation, you will overestimate gene expressions !... unless you decide to count only forward reads or reverse reads.