LOAD TRAINING DATA
For the course "Analyse des Génomes", we need three types of datasets
- The reference sequences that will be used to align sequencing reads (full genome, miRNA, transposons, etc.)
- libraries of sequencing reads from small RNAs (for analysis of piRNAs)
- Librairies of sequencing reads from mRNA (for Gene differential expression analysis)
All these data have been deposited in the storage server Psilo at Sorbonne-Université.
Get data "by URL"¶
As these data are available through a URL (Universal Resource Location) we will use
as before the menu Paste/Fetch Data of the Upload Data menu.
There are other methods to upload data in Galaxy !
- You can transfer data from your local machine (the one where your keyboard is plugged !) to Galaxy
- You can upload data to your Galaxy FTP account and then transfer these data from your Galaxy FTP directory to one of your Galaxy histories.
1. Upload of reference files as a batch of multiple URLs  Programmatic file naming¶
Programmatic file naming¶
As you have already uploaded single files using their url, we are going to use a more powerful procedure which is appropriate when uploading numerous files.
Before all, create a new history by clicking the icon in the history header
and immediately renaming the new history as
References.
- Click the
Upload Databutton at the top-left corner of the Galaxy interface. - This time, Click the
Rule-basedtab ! - Leave Upload data as
Datasetsand Load tabular data fromPasted Table - In the text field
Tabular source data to extract collection files and metadata from, paste the following Tabular source data:
 URLs of references (genome and RNA classes)
URLs of references (genome and RNA classes)
The following list corresponds to the list of genomic features the sequence
of the PLacZ transgene, given in your course manual
https://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.54_FB2023_05/fasta/dmel-all-chromosome-r6.54.fasta.gz dmel-r6.54-fasta
https://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.54_FB2023_05/fasta/dmel-all-miRNA-r6.54.fasta.gz dmel-r6.54-miRNA
https://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.54_FB2023_05/fasta/dmel-all-miscRNA-r6.54.fasta.gz dmel-r6.54-miscRNA
https://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.54_FB2023_05/fasta/dmel-all-tRNA-r6.54.fasta.gz dmel-r6.54-tRNA
https://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.54_FB2023_05/gtf/dmel-all-r6.54.gtf.gz dmel-r6.54-gtf
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_GenomicFeatures/download?path=%2F&files=PLacZ.fasta PLacZ
- Click the
Buildbutton - In the
Build Rules ...pannel that opens, click the and choose
and choose Add/Modify Column Definitions - Click a first time on
Add Definitionand SelectURL. Leave the URL column toA - Click a second time on
Add Definition, selectNameand choose the columnBforName - Now, click the
Applybutton - And to finish the job, click on the dark-blue button
Upload



2. Upload of small RNA sequencing datasets Programmatic dataset naming.¶
- Create a new history using the icon of the history menu, and rename it
Small RNA sequence datasets - Click the
Upload Databutton at the top-left corner of the Galaxy interface. - Click the
Rule-basedtab as we just did with the reference datasets - Leave Upload data as
Datasetsand Load tabular data fromPasted Table - In the text field
Tabular source data to extract collection files and metadata from, paste the following Tabular source data:
 small RNAseq datasets
small RNAseq datasets
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_smallRNAseqData/download?path=%2F&files=ALBA25.fastqsanger.gz WT-ALBA25
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_smallRNAseqData/download?path=%2F&files=ALBA26.fastqsanger.gz WT-ALBA26
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_smallRNAseqData/download?path=%2F&files=ALBA27.fastqsanger.gz WT-ALBA27
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_smallRNAseqData/download?path=%2F&files=ALBA28.fastqsanger.gz GLKD-ALBA28
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_smallRNAseqData/download?path=%2F&files=ALBA29.fastqsanger.gz GLKD-ALBA29
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_smallRNAseqData/download?path=%2F&files=ALBA30.fastqsanger.gz GLKD-ALBA30
- Click the
Buildbutton - In the
Build Rules ...pannel that opened, click the
and choose Add/Modify Column Definitions - Click a first time on
Add Definitionand SelectURL. Leave the URL column toA - Click a second time on
Add Definition, selectNameand choose the columnBforName - Now, click the
Applybutton -
select the Type "fastqsanger.gz" at the bottom of the panel.
 In the menu,
the
In the menu,
the fastqsanger.gzlooks very similar to thefasqcsanger.gzdata type, which is obsolete. The extracmakes a big difference and will put your future jobs in error. Alternatively, you can let Galaxy guess the datatype. Nowadays, it is pretty good at this !
-
To finish the job, click on the dark-blue button
Upload
3. RNAseq datasets (for gene differential expression analysis)¶
- Create a new history in Galaxy and rename it
RNA sequence datasets - Click the
Upload Databutton at the top-left corner of the Galaxy interface. - Click the
Rule-basedtab as we just did with the reference datasets - Leave Upload data as
Datasetsand Load tabular data fromPasted Table - In the text field
Tabular source data to extract collection files and metadata from, paste the following Tabular source data:
 RNAseq datasets
RNAseq datasets
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_RNAseqData/download?path=%2F&files=ALBA4.fastqsanger.gz WT-ALBA4
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_RNAseqData/download?path=%2F&files=ALBA5.fastqsanger.gz WT-ALBA5
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_RNAseqData/download?path=%2F&files=ALBA6.fastqsanger.gz WT-ALBA6
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_RNAseqData/download?path=%2F&files=ALBA1.fastqsanger.gz GLKD-ALBA1
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_RNAseqData/download?path=%2F&files=ALBA2.fastqsanger.gz GLKD-ALBA2
https://psilo.sorbonne-universite.fr/index.php/s/Kdm3_RNAseqData/download?path=%2F&files=ALBA3.fastqsanger.gz GLKD-ALBA3
- Click the
Buildbutton - In the
Build Rules ...pannel that opened, click the
and choose Add/Modify Column Definitions - Click a first time on
Add Definitionand SelectURL. Leave the URL column toA - Click a second time on
Add Definition, selectNameand choose the columnBforName - Click the
Applybutton -
select the Type "fastqsanger.gz" at the bottom of the panel
-
And to finish the job, click on the dark-blue button
Upload
4. Uncompress datasets¶
[Section 4 should be optionnal, see with Stéphane, I do not think it is
necessary, but he's the boss !]
At this stage, we have uploaded small RNA and RNA sequencing datasets as fastqsanger.gz.
To simplify the subsequent analyzes we are going to uncompress all these datasets, whose
datatype will therefore become fastqsanger.
Procedure for a single dataset¶
- Go to your
small RNA input datasetshistory (or whatever you named it). - Click on the pencil icon
 of the first dataset.
of the first dataset. - Click on the tab
datatype .
. -
In the panel
Convert to Datatype, selectfastqsanger (using 'Convert compressed file to uncompressed.')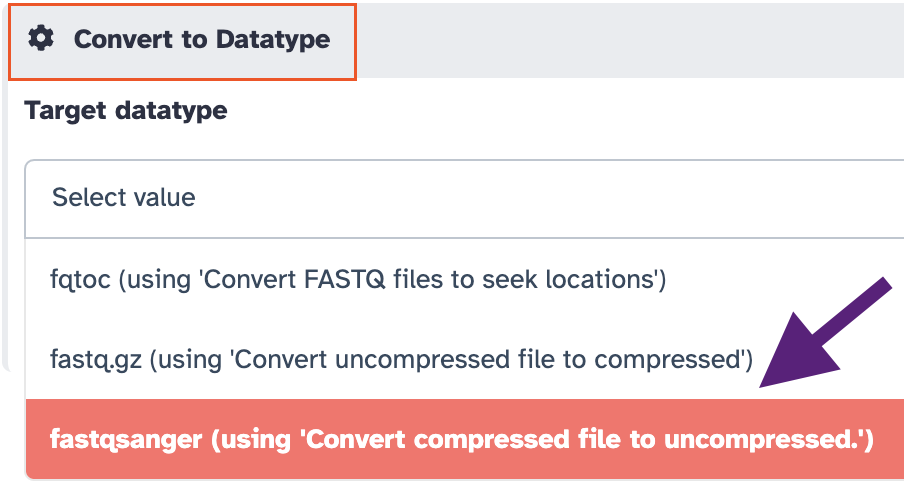
Why NOT using the panel
 ?
?- Let's imagine a Galaxy dataset whose name is
Hamlet - the content of this dataset is:
- Would you agree that the
datatypeof this dataset isenglish? I think so. - Let's put it all together in the form of:
Now, what if you change the
This does not seem correct ! Do you aggree ?Datatypeof this dataset fromenglishtofrenchusing theAssign Datatypepanel? This →If you
It is looking better, isn't it ?Convertinstead this dataset fromenglishtofrench, you will have This →In contrast, if your starting dataset was as this:
There, you would "just"Assignthe Datatype of the dataset fromenglishtofrenchand get: - Let's imagine a Galaxy dataset whose name is
-
Click on

→ A new dataset is created. During the decompression job, its name looks like
5: Convert compressed file to uncompressed. on data 1. But when the job finishes, the
name of the dataset changes to more self-explanatory: 5: GRH-103 uncompressed.
Repeat the same procedure for every small RNAseq dataset.¶
Repeat the same procedure for every RNAseq dataset.¶
Naturally, you can launch as many jobs as you need in the same time
When all datasets are decompressed¶
- Delete the compressed datasets (by clicking on the cross icon of datasets).
- Rename the uncompressed datasets by removing the
uncompressedsuffix. -
Purge the deleted datasets. This is done by clicking the wheel icon of the top history menu, and selecting
Purge Deleted Datasetsin the Datasets Actions section.
-
If you do not perform this last action, the deleted datasets remain on your
instance disk !
5. Dataset collections  ¶
¶
We are going to organize our various datasets using an additional structure layer: the Galaxy Collection.
A Galaxy Collection is a container object which is convenient to treat together multiple equivalent datasets, such as a list of sequencing datasets, of text labels, of fasta sequences, etc.
A. Making collections of RNA sequence datasets.¶
Collections are particularly useful for RNAseq datasets,since these datasets often come
as replicates which can be grouped upon a label. Your training is indeed a good example of
that, since you are provided with 3 WT datasets (ALBA4, 5 and 6) and 3 GLKD datasets
(ALBA1, 2 and 3).
- Navigate to you
RNAseq inputshistory (or whatever you named it) and click the upper left small check box at the top of the dataset stack
You see that check boxes appear for each dataset of the history
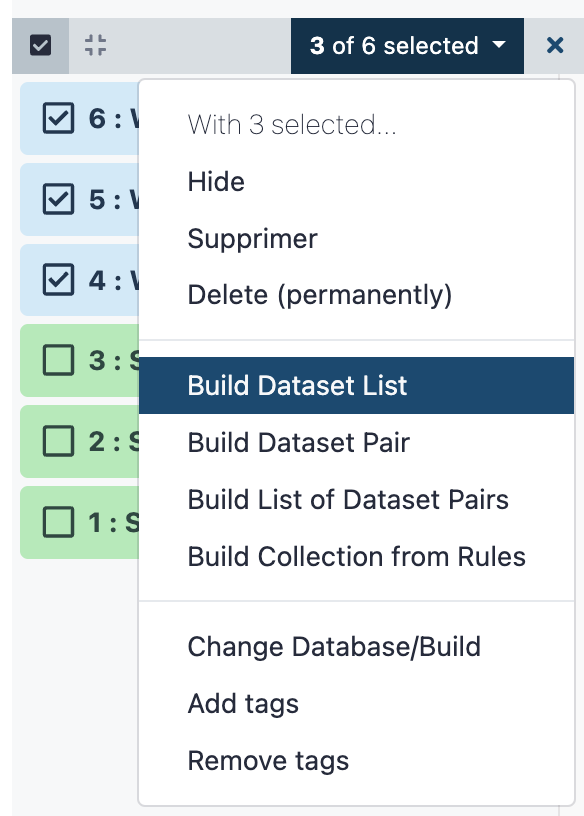
- Check the 3 RNA datasets
WT(-ALBA4, 5 and 6) - In the menu
3 of 6 selected(also in the top area of the history), selectBuild Dataset List
- In the pop-up panel, just type
WTin the fieldName: Enter a name for your new collection - Reorganize the datasets order by clicking the
alphabetic sortingicon. -
Press the button
Create Collection -
Repeat exactly the same operations for the 3 remaining datasets
GLKD(-ALBA1, 2 and 3) - When you are done with the creation of collection, you can uncheck the upper left small check box
What do you see when you click on name of the new dataset collections ?
You see the content of the collection, with datasets identified with original names.
Click on the << History link, to come back to the normal history view.
what do you see if you click the crossed eye icon at the right corner  ?
?
You see the actual datasets contained in the Collection. If you click on unhide for
each of these datasets, you will actually see permanently both the container collection and the contained
datasets !