
STAR alignments¶
Let's test another aligner, STAR, in another new history. It will later be possible to compare the performance between STAR and HISAT2.
Before anything, let's use some nice features of Galaxy to manipulate quickly datasets and histories.
- Go, or stay, to the history
HISAT alignments - Click the upper right wheel icon at the top of the history stack and select
Copy datasetsorCopier des jeux de donnéesif your interface is french.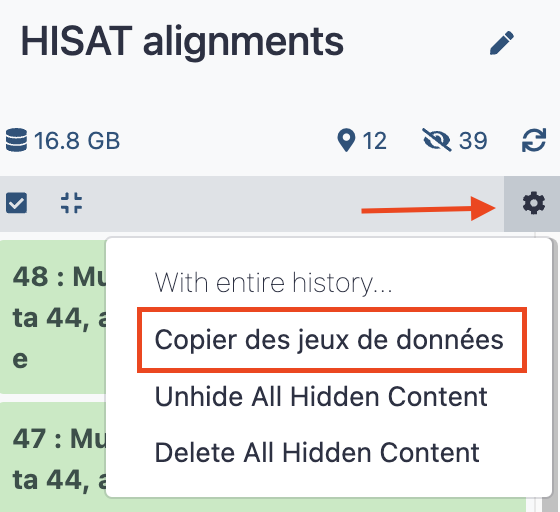
- In the pop up panel, select the three dataset collection that we already built in the
history. In the right part of the panel (
Destination History), fill the empty fieldNew history namedwithSTAR Alignments, as shown bellow, and last, click theCopy History Itemsbutton.

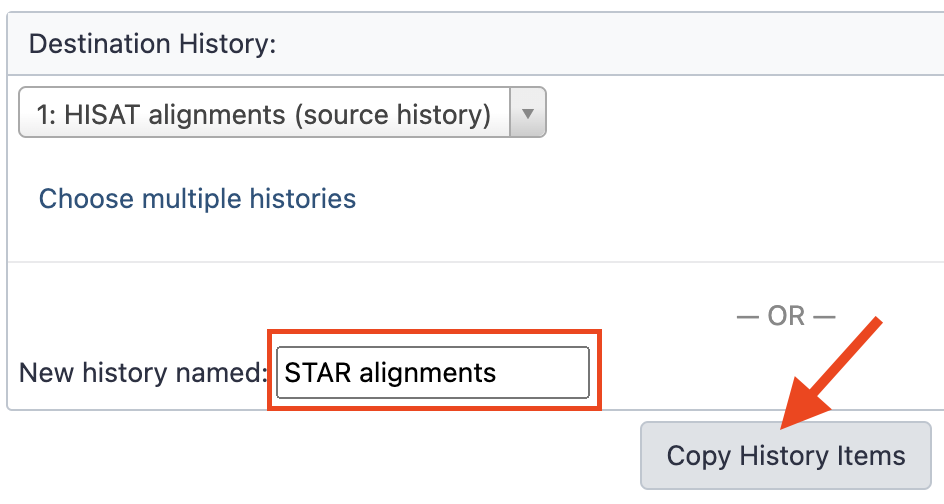
- Navigate directly to the newly created history by clicking the link 3 datasets copied to 1 history: STAR alignments.
- You could also have copied the GTF file which will also required for STAR alignment, button for practicing purposes, let's get this file from the data library. We assume that you now know how to do this !
- This makes our new history
STAR alignmentslooking as follows:
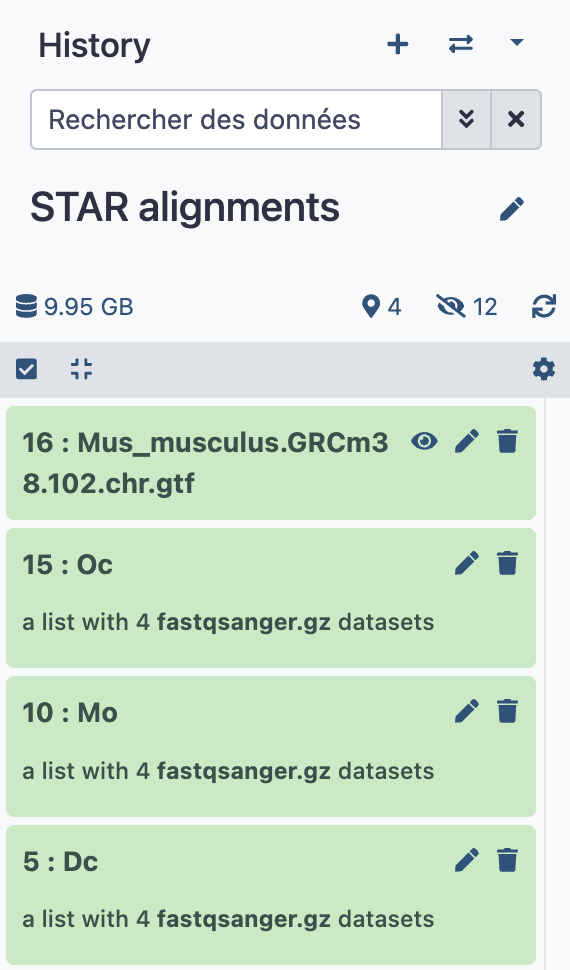
 RNA STAR tool¶
RNA STAR tool¶
![]() RNA STAR settings
RNA STAR settings
-
Single-end or paired-end reads
→ Single-end
-
RNA-Seq FASTQ/FASTA file
→ select the collection icon and then the collection
5: Dc -
Custom or built-in reference genome
→ Use a built-in index
-
Reference genome with or without an annotation
→ use genome reference without builtin gene-model but provide a gtf
-
Select reference genome
→ GRCm38_w/o_GTF
-
Gene model (gff3,gtf) file for splice junctions
→ Mus_musculus.GRCm38.102.chr.gtf
-
In
Output filter criteria, Exclude the following records from the BAM output→ check Select all
-
Leave any other setting as is and Press
Execute!
The tool will run during several minutes, generating four new dataset collections, whose name is self-explanatory. Take benefit of the run time, to rename at least 3 of these collections with more meaningful names:
RNA STAR on collection 5: log→Dc STAR logRNA STAR on collection 5: mapped.bam→Dc RNA STAR mapped.bamRNA STAR on collection 5: reads per gene→Dc nbre of reads per gene (STAR)
 Reminder: we understand it is a bit borring to rename datasets but these
renaming operations are essential to the readibility of your histories.
Reminder: we understand it is a bit borring to rename datasets but these
renaming operations are essential to the readibility of your histories.
Re-run the RNA STAR tool as you learned before with HISAT for the collections:¶
-
10: Mo -
15: Oc - ... And do not forget to rename your collections accordingly

About RNA STAR splice junctions.bed collection
These datasets are useful if you seek to find new splicing events and describe them.
Since here, we are performing annotation-guided RNAseq analysis, these collections of datasets are not useful and you can trash them !
Mapping statistics with MultiQC tool¶
![]() MultiQC settings
MultiQC settings
- 1: Results
-
Which tool was used generate logs?
→ STAR
-
Click "Insert STAR output"
-
Type of STAR output?
→ Log
-
STAR log output
→ Click first the collection icon
→ Select the 3 collections
Dc,MoandOc RNA STAR log, holding down the Cmd key -
Leave the other settings as is
- Press
Execute!
When MultiQC has run, look at the aggregated mapping statistics by clicking the eye icon
of the dataset MultiQC on data 46, data 44, and others: Webpage