lumpy analysis
Now we are going to analyse the alignments generated by BWA, using the tool lumpy-sv.
The lumpy-sv tool analyses the alignments in the BAM file and searches for
-
split alignments
reads whose 5' and 3' parts map to non-contiguous regions
-
discordant alignments of read pairs
Either one mate maps to one chromosome and the other mate maps to another chromosome, or the distance between the two mates is beyond what is statistically expected (the library fragment size in average). For insertions, the distance increases, for deletions, the distance decreases
From this parsing, lumpy then constructs models of break points that can explain its findings, and report these models in a vcf file (vcf stands for variant calling format), with statistical significance, number of evidences founds, etc...
-
launch the lumpy-sv tool
You can use the search bar at the top of the left-hand column and type
lumpy -
Adapt the tool parameters:
lumpy-sv find structural variants (Galaxy Version 1.1.0)
- input(s):
One Sample(because we are not comparing the alignments with a reference alignment) - One BAM alignment file produced by BWA-mem:
Map with BWA-MEM on collection 5 (mapped reads in BAM format)
You need to toggle the dataset collection mode (folder icon) to see the collection (arrow in the screen shot below).
- read length:
151 - Sequencing method:
Paired-end sequencing - variant calling format:
vcf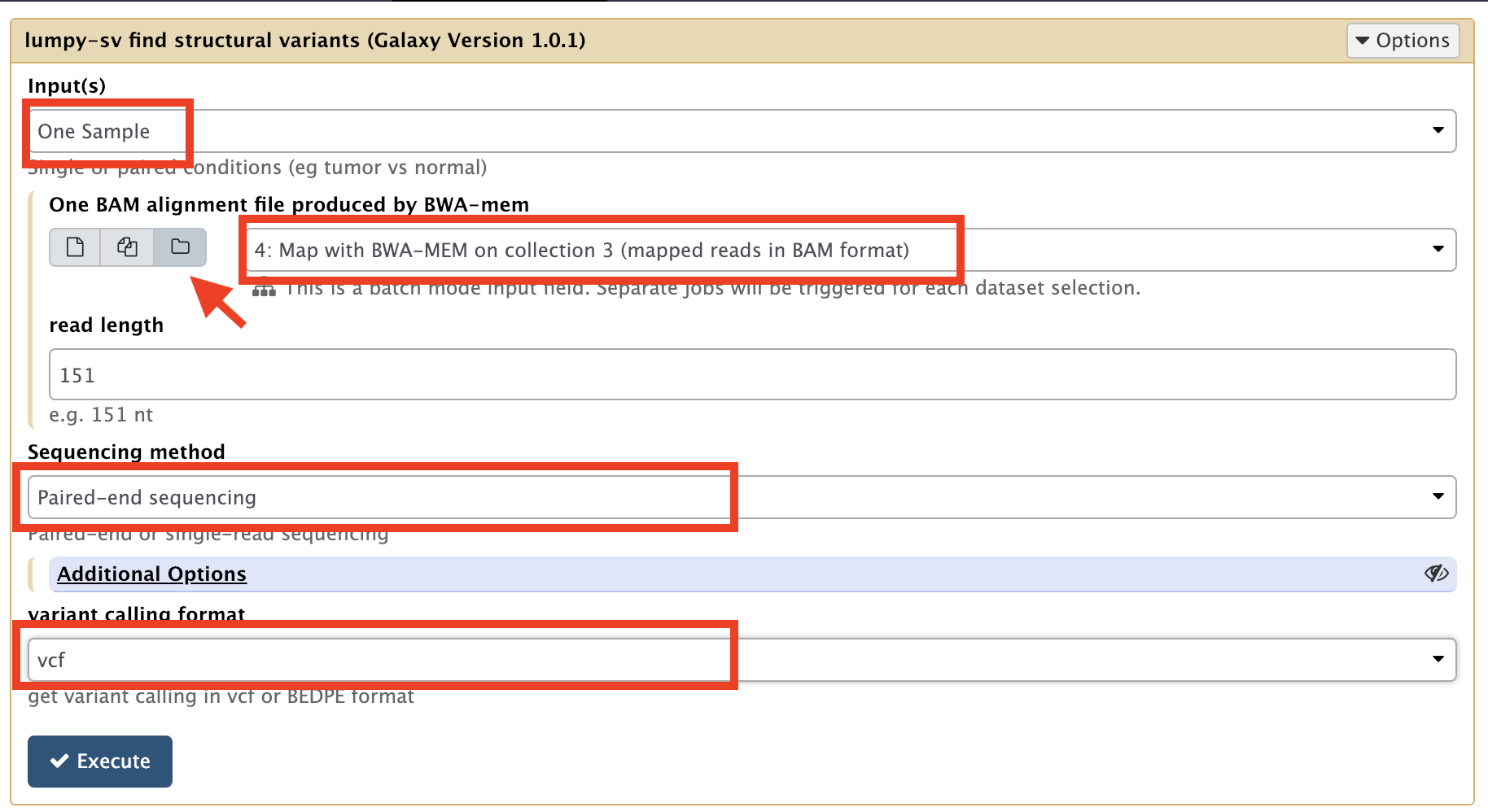
- input(s):
-
Click the Execute button to run the tool
-
Look at the vcf returned by lumpy-sv.
lumpy-sv vcf output
lumpy-svoutputs variations as a suite of single lines (for deletions, insertion, or SNPs), or as a suite of line pairs, for translocation (one line for the translocation, the other for the reciprocal translocation). Note that sometimes, there is evidence for one translocation, but not for the reciprocal event. Look at the ID column: a pair of translocation event model will have for instance IDs 2_1 and 2_2, respectively.However, the lumpy vcf format is not suitable for visualisation in a genome browser such as the UCSC genome browser.