Align reads to the human genome
We are going to use the BWA-mem aligner to align reads to the human genome, version hg38.
-
Select the BWA-MEM tool in the Galaxy tool bar

-
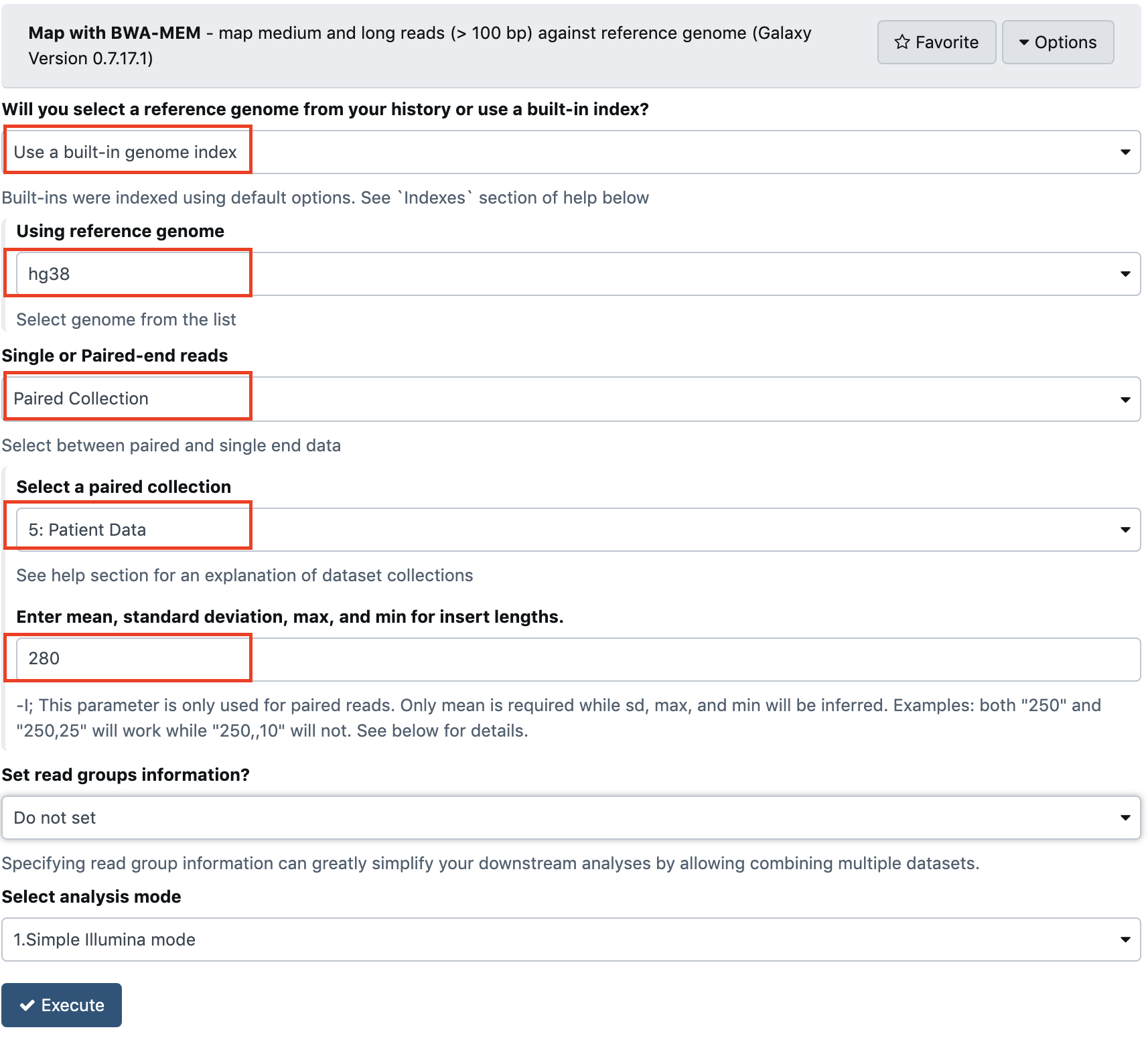
Fill carefully the tool form
Map with BWA-MEM - map medium and long reads (> 100 bp) against reference genome (Galaxy Version 0.7.17.1)
- Will you select a reference genome from your history or use a built-in index?:
Use a built-in genome index - Single or Paired-end reads:
Paired Collection- Select a paired collection:
A or B collection - Enter mean, standard deviation, max, and min for insert lengths.:
280
- Select a paired collection:
- Set read groups information?:
Do not set - Select analysis mode:
1. Simple Illumina mode
This should give you a form similar to:
- Will you select a reference genome from your history or use a built-in index?:
-
Click the
Executebutton