Run workflow
In this use case, we are going to
- Upload a workflow description files in your Galaxy server instance
- Visualise this workflow and its tools
- Eventually run the workflow on input data obtained from a remote public repository.
1. Upload workflow description file (.ga)¶
- Click the
workflowmenu - Click the "Upload or import workflow" button at the top right
-
In the
Galaxy workflow URL:field, paste the url of the workflow file:Note that this file is in the artbio/AnalyseGenome repository where material for this training is hostedhttps://raw.githubusercontent.com/ARTbio/AnalyseGenome/main/Exercises/Galaxy-Workflow-Extract_canonical_transposons_fasta.ga -
Click on the
Importbutton
Note
Alternatively, you could upload the workflow files from you computer instead of uploading them by URL
- the
Workflowmenu should now look like :
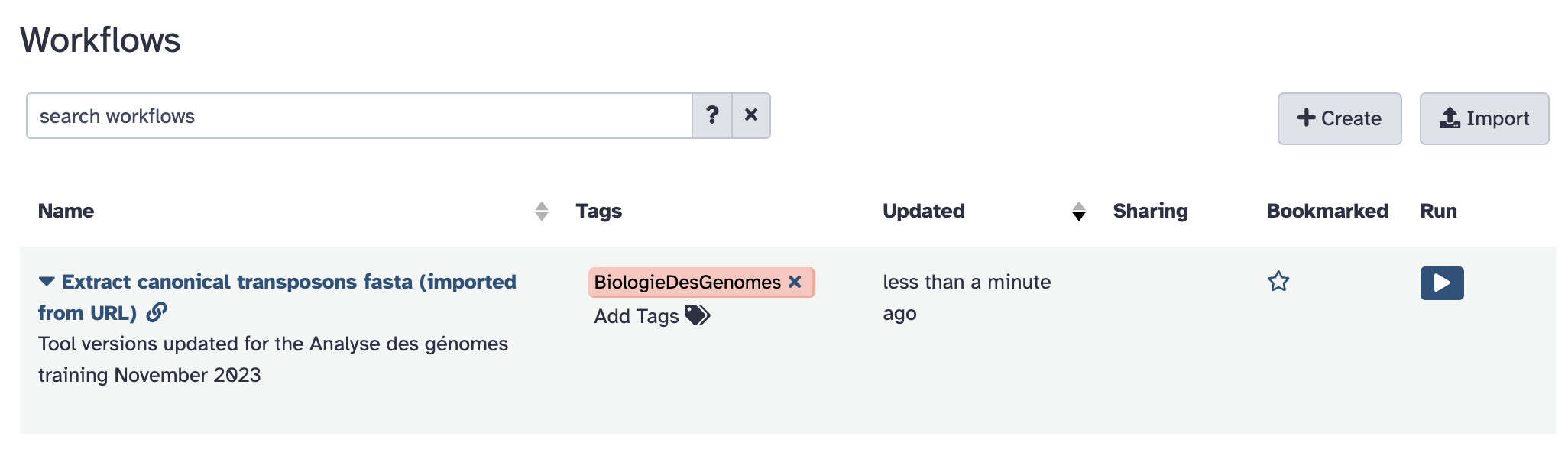
- Click the workflow
Extract canonical transposons fasta (imported from URL)and theEditoption
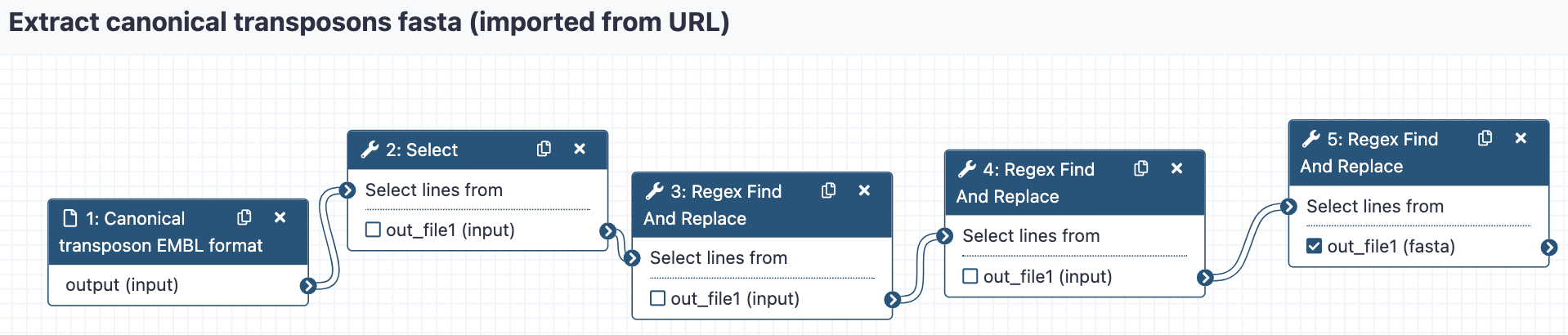
We can go through the various steps of the workflows and figure out what they are doing.
This workflow performs a series of find-and-replace text manipulations, starting
from input data that has been tagged transposon_set_embl.txt and producing a new text
dataset that is renamed Canonical_transposons.fa.
4. Retrieve the transposon_set_embl.txt dataset¶
- Create a new history and name it
workflow test -
import the dataset
- Either using the
Paste/Fetch datamode of the upload manager (the small bottom-top arrow icon at the top left of the Galaxy interface). Copy the URLin the open field and click thehttps://raw.githubusercontent.com/bergmanlab/drosophila-transposons/master/releases/transposon_sequence_set_v9.5.embl.txtStartbutton. -
OR, remembering that you just did that few minutes ago !
Thus, you can use the copy datasets function which allow to copy datasets from history to history !
How to copy a dataset from an existing Galaxy history
- Click the wheel icon at the top of your history right bar
 - Select
- Select EMBL to Fasta conversionin theSource Historyleft panel - Check the item1in the list (should betransposon_sequence_set_v9.5.embl.txt) - Verify that the history in theDestination Historyright panel isworkflow test(should be by default, otherwise change it) - Click theCopy History Itemsbutton - Observe the dataset showing up in yourtest workflowhistory !
- Either using the
-
have a close look at the dataset
5. Run the workflow¶
- Click on the workflow menu
- Click the Run option of the workflow (the
 to the right hand side)
to the right hand side) - Select the appropriate dataset (should be only one already selected)
- And Click the
Run workflow - Look at datasets in the history turning from grey to yellow to green and eventually getting hidden.
6. Check result¶
You may check that the generated dataset is identical to the one generated with the tool
embl2fa using the tool differences between two files
7. Goody for you: an exemple of workflow to treat complex data table¶
As this is a goody, we put here the key steps to run the workflow
- Workflow URL
- 2 tools must be installed in your Galaxy instance to get the workflow running:
add_column_headerscolumn_maker- Input table to be parsed/transformed